化学結合にそれぞれ理想的な結合長や結合角があります。それらから外れると、元に戻そうと斥力や引力が働き、ポテンシャルエネルギーは高くなります。分子力学法では、一度、化学結合は電子間の相互作用であるという概念を忘れ、原子と原子は化学結合という名前のバネで結ばれているという大胆な過程の基に計算を行います。
下には、理想的結合長A-B、および増大します。それぞれのポテンシャルエネルギーは結合長、或いは結合角の関数ということができます。分子力学法では、それらに一つ一つのポテンシャルエネルギーカーブを実験データーや、分子軌道計算結果に基づきパラメータとして作成し、理想値からのずれによって生じるさまざまなポテンシャルエネルギーの蓄積を計算する方法です。
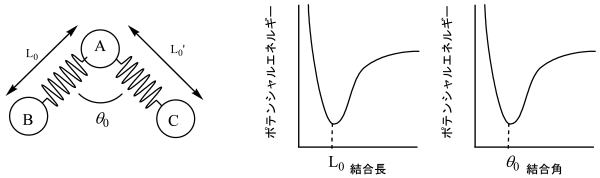
さらに結合角回転や秘訣合成の相互作用のファンデルワールス(VDW)相互作用、静電相互作用などを考慮して、分子力学法では分子の持つポテンシャルエネルギーを
として表します。これを分子力場 (Molecular Force Field) といいます。
考慮する項目の取り方は力場によって異なります。パラメータ作成には、実験値のみでなくab initio法で求めた理論値も用いられます。分子力学法では、全てパラメータが分子の持つエネルギーを決定します。従って、パラメータの精度が低ければ意味がありません。アセトアルデヒドのC1-C2結合の結合距離が1.053Åであるのに対し、ベンズアルデヒドのそれは、1.483Åと同じ炭素-炭素結合でも周りの環境によって異なるように実際のプログラムでは注目する結合の周辺環境も考慮し適切なパラメータを選ぶ工夫がなされています。パラメータの作成にはさまざまな工夫が行われMM1, MM2, SYBYL, MMFFなどの分子力場が作成されました。
パラメータの基になった化合物群では驚く程高い再現性を示します。逆にパラメータ作成時に考慮されていない官能基をもつ分子群では、信頼性の低いものとなってしまいます。従って、計算しようとする分子に適切な分子力場を用いる必要があります。
分子力学法では、分子軌道計算に比べ計算の負荷が極めて少なく、数万の分子量即ち、タンパク質レベルまで計算可能です。
厳密に考えると分子力学法では、全ての官能基でパラメータが定義されている分子種のみ適用可能となります。たとえば、MM2ではシクロプロパンは計算できません。そのまま実行すれば、エラーで終了してしまいます。計算する場合、シクロプロパンのオプションを追加する必要がありました。最近のプログラムではパラメータを持ち合わせない場合は、自動的に類似のパラメータを流用する、或いは量子力学法に基づき作成するなどして多くの分子を計算できるようになっています。しかし、パラメータの流用などコンピューターが行ったそれぞれに対する措置が適切でなければ、計算結果に意味はありません。たとえばChem3Dに使用されているMM2では計算の実行を優先し、オリジナルのMM2とくらべ、計算可能な分子種は格段に広くなっています。裏を返せば、パラメータの流用などが行われているはずなので、計算結果の評価には、まずその詳細を調べることが必要です。たとえば硫黄に置換した糖の立体配座を硫黄原子を評価するパラメータを持たず、可能なパラメータを発生させる分子力場を使用したとします。このとC-S結合のパラメータが無いためC-Oのそれを流用するプログラムではモデルの元素記号だけが「S」となった普通の糖の立体配座を計算することになり、通常糖と硫黄置換糖とを比較しても、全く同じものを比較することになってしまいます。分子力場計算ではパラメータの適用範囲を常に意識する必要があるといえます。
分子力学法では、分子の安定構造における原子の座標を得るという目的以外の要素は考慮されていないため、スペクトルなどの物理特性の予想は出来ません。
分子力場として、以下のものがよく知られています。
MM2、MM3、MM4 (Molecular Mechanics program 2 or 3 or 4)
Allingerによって作成された分子力場で、計算を主としない論文でも多く用いられている方法です。オリジナルは、MOPACと同様にCUI(command line user interface)で入力ファイルの作成、計算結果の解析を行っていましたが、Chem3D(Cambridge Software)などに組み込まれるようになり、モニター画面上でGUI(Graphic User Interface)操作で入力ファイル作成、計算結果の解析が出来るようになっています。HyperChem(Hypercube, Inc)ではMM+という力場が使用できます。
MMFF94 (Merck Molecular Force Field 94)
メルク社によってMM3をベースに開発された力場で、極性の高い官能基を持つ分子でも良い再現性を示します。パッケージプログラムとしてはSpartan シリーズ(Wave Function)に組み込まれています。MMFF94については力場の詳細についても公開されています。
AMBER (Assisted Model Building and Energy Refinement)
AMBER(Assisted Model Building with Energy Refinement)は分子動力学シミュレーションソフトウェアのことですが、amberという力場はHyperChem(Hypercube, Inc)に採用されています。
CHARMM (Chemistry at Harvard Macromolecular Mechanics)
SYBYL
これら分子力場のパラメータは、タンパク質、ペプチドなど生体分子に焦点をあてて作成されています。CHARMはDiscovery Studio (Accelrys)、SYBYLはSpartanシリーズ (Wave Function)で利用可能です。