ペプチドの計算
ペプチドを計算する場合、どのような方法で計算するとよいのであろうか?
こんな単純な疑問から手持ちのプログラムを無手勝流で計算してみた。
計算に用いたのはアラニンのペンタマー。
1.Chem3Dにパッケージされているプログラム
MM2
Chem3Dには古くからMM2がパッケージされている。本来のMM2ではちょっと変わった官能基があると「パラメータが無い。」といって止まってしまう。感覚的には計算できる分子のほうが少ないといった感じである。
ところがChem3DのMM2は何でも計算できる。ケンブリッジソフトのホームページによればChem3D Pro, Chem3D Ultraでは、パラメータがなくても、”Chem3D can guess at missing parameters”と書いてある。
つまりソフトウェアが勝手にパラメータを作っているということである。
計算が開始されるのはボタンを押して直後なので、分子軌道法でパラメータを作っているというものではないらしい。
すなわち、似た構造のパラメータを流用しているということらしい。
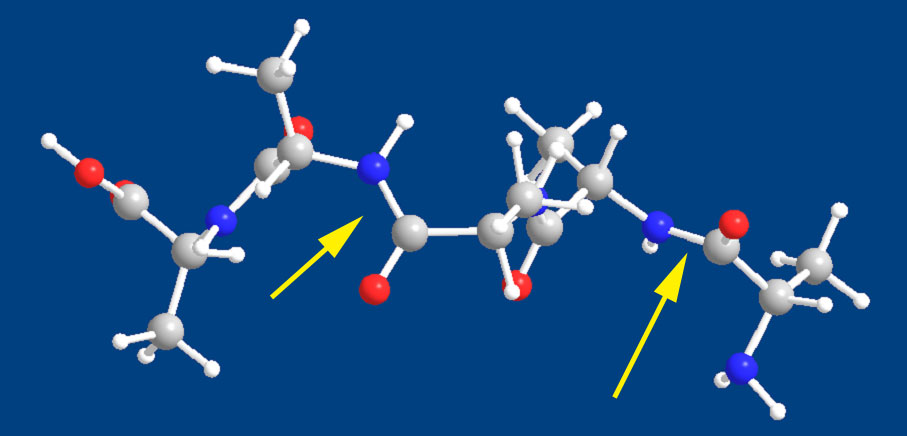
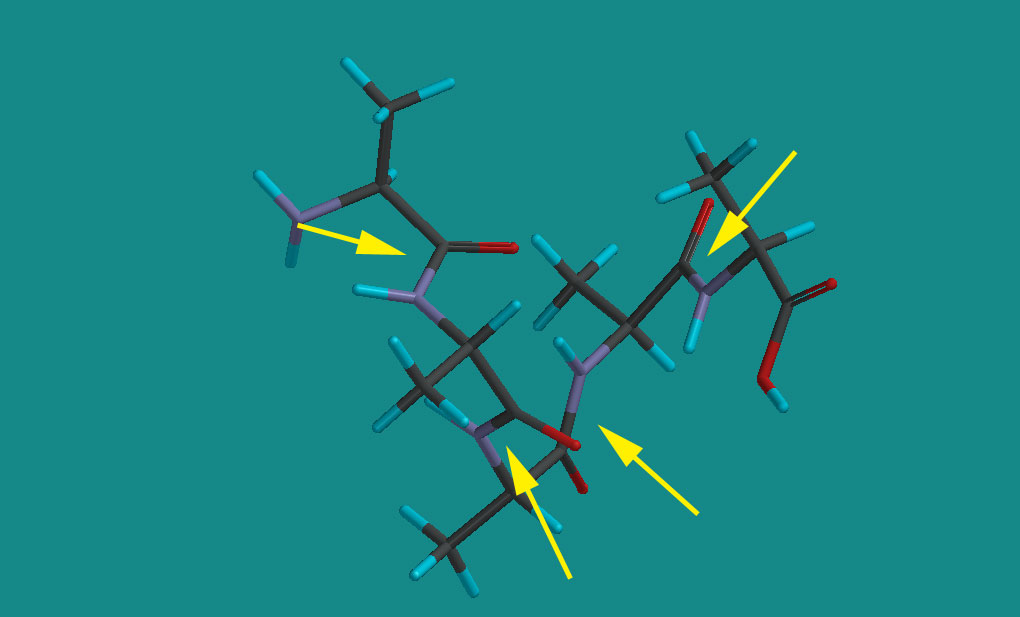
それでも計算で、真実に近い構造さえ出してくれれば何も問題ないわけである。ということでアラニン5量体を計算してみた。
上に示した計算結果を見ても一目瞭然で、この計算はダメである。ペプチドのカルボニルとアミド水素とは平面に並ぶはずなのに大きく歪んでいる。Chem3DのMM2はペプチドの計算はできないと判断すべきであろう。
MOPAC
Chem3DにはCS MOPAC Proという正式なMOPACがパッケージされている。すなわちMOPACのキーワードがそのまま使える。ペプチドの計算ではキーワード"MMOK"を加えてアミド水素を分子力学補正する必要があることは言うまでもない。最近のパソコンの能力はすばらしく、これくらいの分子だと数分で計算が終了してしまう。
AM1
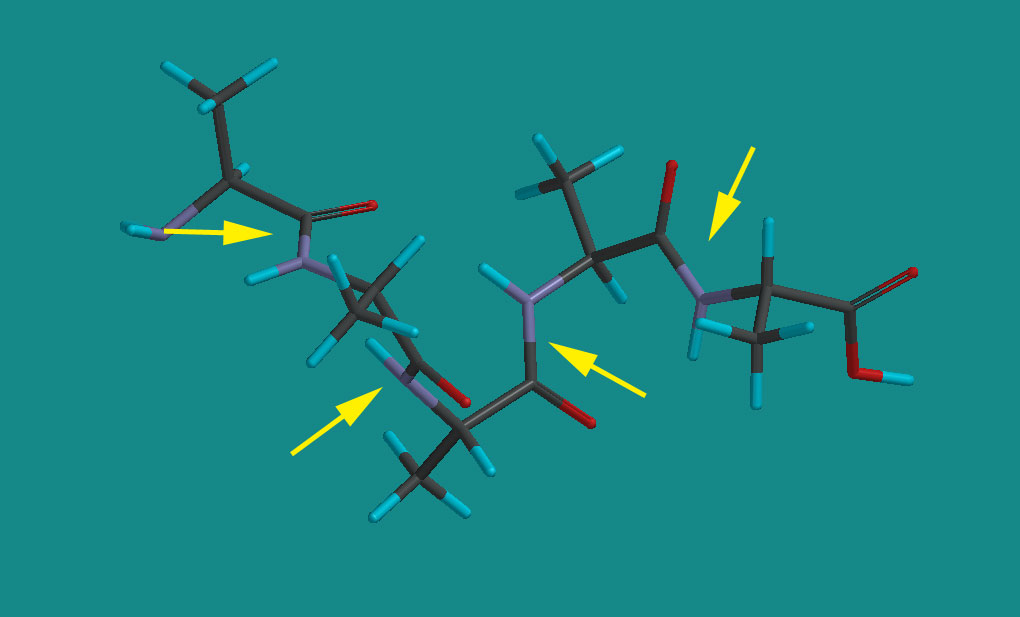
まずAM1について計算してみた。
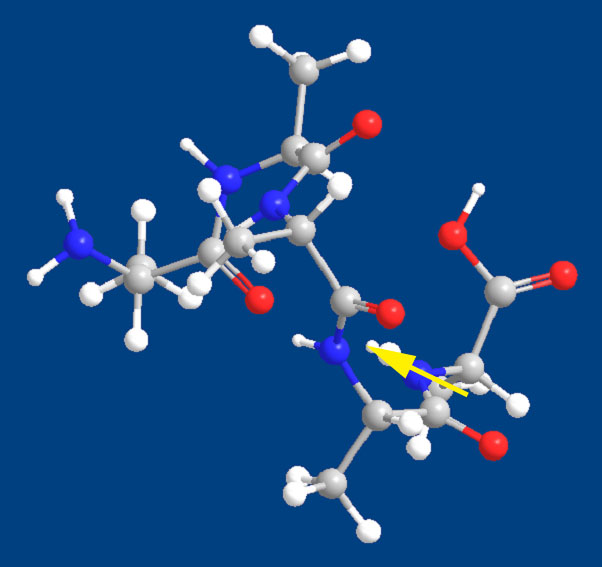
計算結果はアミドをほぼ完全に平面に計算しており、この点からは、ペプチド計算に使えそうだ。
PM3
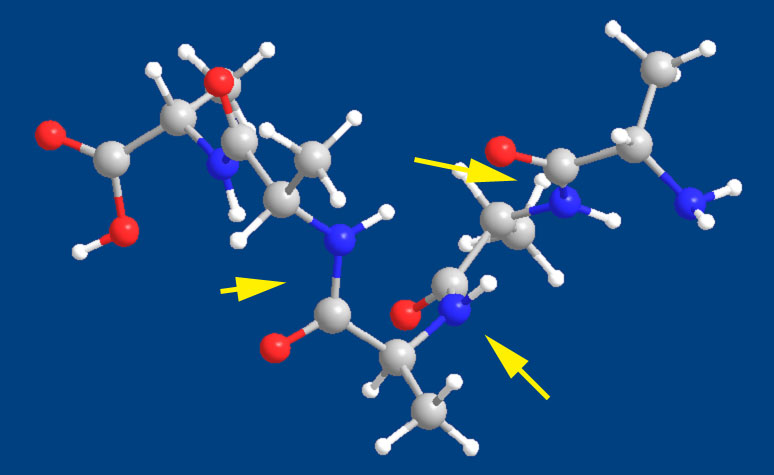
次にPM3をやってみた。
これもペプチド結合に関してはよさそうだ。
示した図ではアングルが変わってしまって判りにくいが、AM1の結果と類似した構造を与えている。
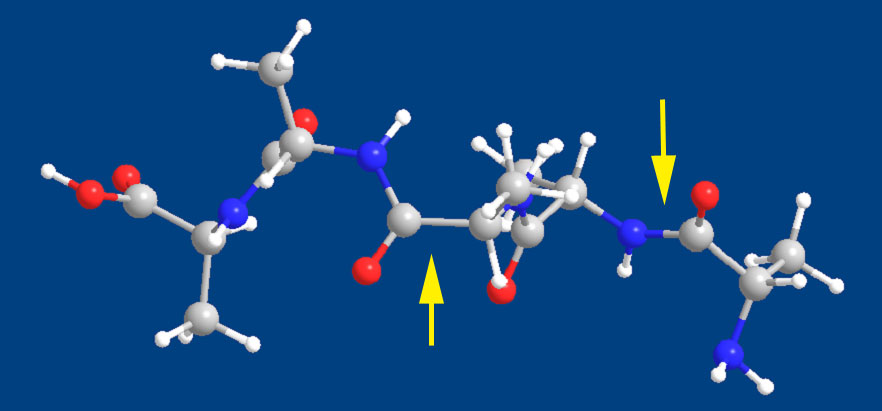
MINDO3
MINDO3もペプチド部分では問題がなさそうだ。
この計算は、PM3の計算結果を初期座標にしたにもかかわらず、全体の配座が大きく変化した。(時間も結構かかった。)すなわち、AM1、PM3とは大きく違う配座を最安定とする可能性が高いといえる。
MINDO
MINDO法もアミド結合については問題がなさそうだ。この計算も先のMINDO3での計算と同じPM3の結果を初期配座として最適化したが、配座が大きく変化し、むしろMINDO3の結果とほぼ同じ配座を示した。名前も似ているが用いている関数もパラメータも似ているのであろう。MINDO法では原子間反発を過大評価するのが欠点であったらしい。
GAMESS
GAMESS(US)は、Iowa州立大のGordon,Moscow州立大のGranovsky,によって開発された分子軌道計算プログラムである。Chem3DでもGAMESSは利用可能となっているが、ケンブリッジソフトのCDからはインストールされない。http://www.msg.ameslab.gov/からダウンロードする必要があった。
ダウンロード時に自分のメールアドレスを登録、しばらくすると解凍のパスワードが送られてきた。
インストール時、パソコンの設定を調べ自動的にインストールしてくれたのはいいが、このとき、ものすごーく時間がかかった。安全のためパソコンにはノートンユーティリティーを入れているが、これがいけないのかと思い、通信を遮断、Auto-Protectをオフ、さらにノートンユーティリティーをログオフしてやっとのことインストールできた。ノートンユーティリティーそのものがウィルスだと感じる今日この頃、でも、入れないわけにも行かず、速くなったパソコンをわざわざ遅くして使っている気がする。
下の図を見ても判るように、設定は極めて細かい。学生時代、量子化学をサボった自分にはさっぱりわからない。
とりあえず、デフォルトの状態でAM1、PM3の二つをやってみたが、アミド部分は平面にならなかった。これはAM1、PM3というハミルトニアン自身が持つものらしい。中身がわからないので意味が無いが、Wave functionについて Open Shellを指定して計算したところアミド部分は平面になった。でも意味がわからない状態には変わりがなく、この辺でやめることにした。

2. SPARTANにパッケージされているプログラム
MMFF
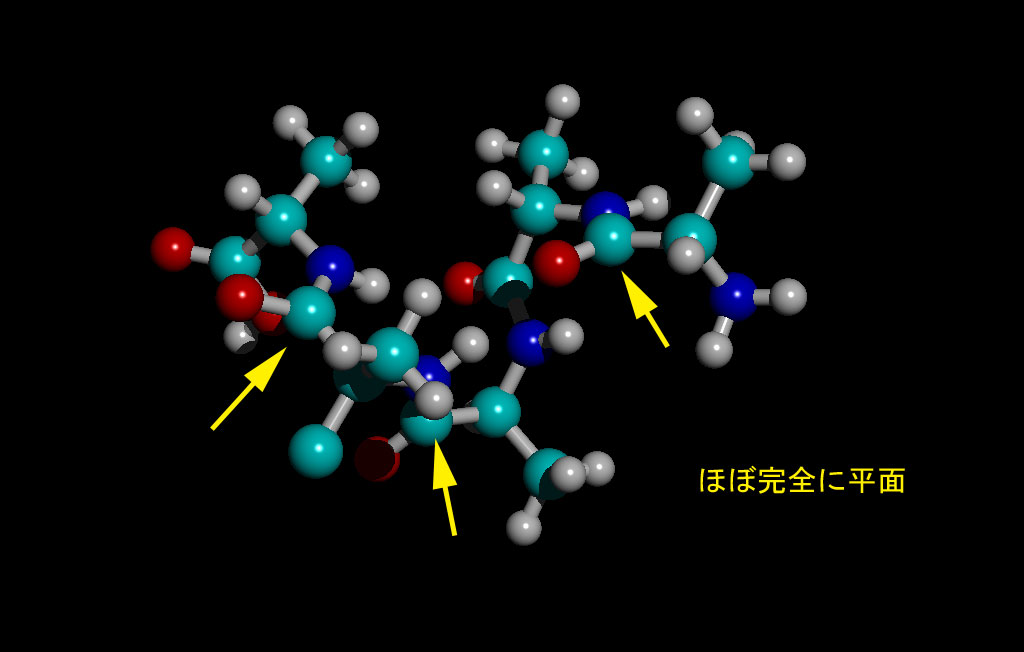
アミドは平面に表現された。SPARTANで分子力学法を選択した場合、デフォルトがこの力場である。開発はメルク。
私自身はこの力場は優秀だと勝手に思っている。
SYBYL
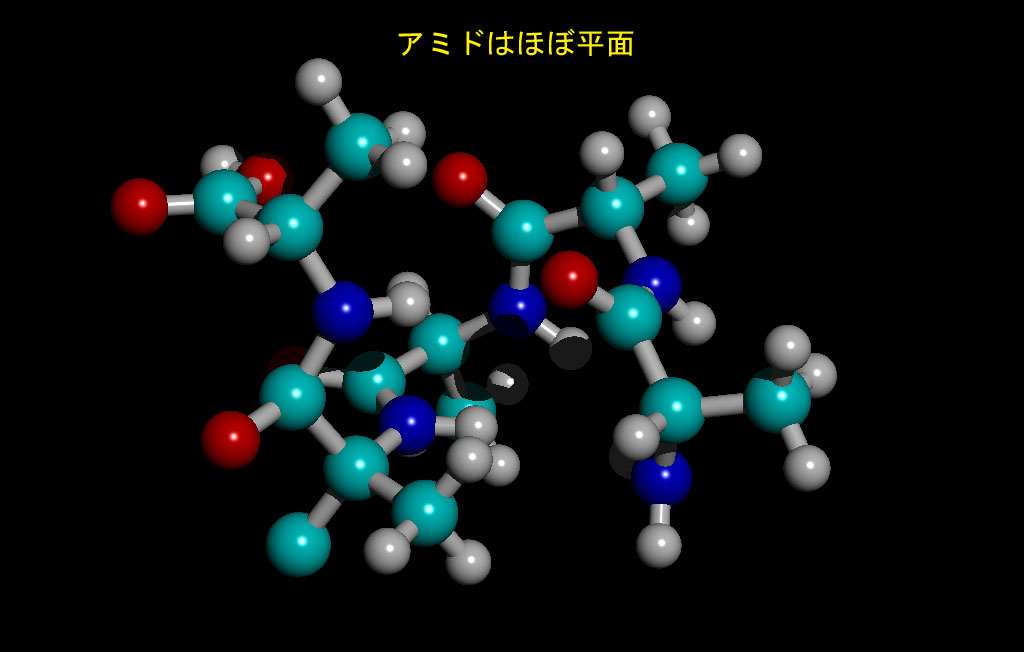
アミドは平面に再現された。得られた配座もMMFFの結果と近いが、最適化のiterationの回数が多く、MMFFでの最適化構造を初期座標にしたにもかかわらずデフォルトではエラーが起きた。計算条件設定時にiterationの最大値を大きくする必要がある模様。
TRIPOS社よって開発されたこの力場は、タンパク質やタンパク質-低分子のドッキング実験に威力を発揮するとホームページに書かれているのでペプチドの再現には裏が取れていると考えてよいであろう。

AM1
Spartanの半経験的分子軌道法にはAM1、PM3とMINDOがあり、MOPACとほぼ同じ内容である。
半経験法においてデフォルトではAM1が選択されている。アミド結合については問題がなさそうだ。計算ではSYBYLの計算結果を初期配座に用いたが、小さなところのみの違いで全体の形が大きく変化したということは無かった。
PM3
SpartanのPM3ではアミド結合の二面角は180度とならなかった。キーワードに”MMOK"を入れて再計算をしてみたが構造は全く変化せず、Spartanではこのキーワードは受け付けないらしい。マニュアルのどこを見ても分子力学補正という言葉は見当たらなかった。
同じハミルトニアンでも計算結果が異なるのは以外であった。もっとも、MOPACでも”MMOK"をキーワードに入れなければ同じ計算結果になったのかもしれない。
この計算結果、ずれているといってもChem3DのMM2ほどではなく、分子全体のプロファイルもAM1の結果と大きな違いは無いので全く意味の無い計算かと言うとそういうわけではないらしい。
いずれにしてもペプチドの計算ではSpartanのPM3は避けたほうが賢明かもしれない。

ab initio法
この計算は時間がかかるのでやっていない。しかし、The Spartan Molecular Databaseにアラニン3量体の3-21G、6-31G*, B3LYP/6-31Gの計算結果が登録されていた。もちろんペプチド結合は完全に平面であった。
B3LYP/6-31Gの座標を初期座標としてほかの計算法をほかの計算法を試みたところMMFFで構造変化が最も小さいように感じられた。この情報は重要である。
Hyperchemにパッケージされているプログラム。
MM+
HyperChemでは計算の細かなセッティングが可能であるが、初心者が計算手法(エネルギーの落とし方??)のパラメータをいじると何が起きるのか判らないのでデフォルトで計算した。
HyperChemのMM+ではアミド結合はきれいに平面に表現された。
AMBER
AMBERはタンパクの構造計算で多く用いられている。したがってペプチドの計算では問題が無いであろうと考えた。
実際、アミド結合は平面で計算された。さすがである。
ただし、別の計算で非天然のアミノ酸残基を有する化合物を計算したら、その部分ではアミド結合は平面どころかズタズタになった。
AMBERは天然型アミノ酸によるペプチド専用と考えたほうが安全であろう。AMBERも正規版では天然型と一部の非天然アミノ酸以外はパラメータが無いといって計算がとまるらしい。
OPLS
OPLS(Optimized Potentials for Liquid Simulations)はその名のごとく水溶液中の分子の挙動を再現することを目的に作られた力場である。生体分子の計算では定評のある分子力場らしい。
アミドは問題なく平面に表現された。
HyperChemでももちろん分子起動計算ができる利用可能なハミルトニアンもextend Huckel, CINDO, INDO, MINDO, MINDO3, MINDO/d, AM1, PM3, ZINDO/1, ZINDO/Sと豊富である。ただこれらの違いを勉強していないので、これらについて調べることは今後の課題とした。