計算方法の選択??
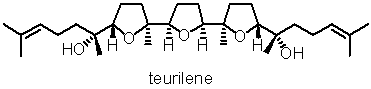
テウリレンを例として
学生時代にテウリレンという8つの不斉炭素にもかかわらず、分子全体としてはメソ体という変わった分子の合成をやっていた。この分子はX線で構造が確かめられたが、結晶中ではヘアピンのように折りたたまれた構造をとっていた。
この分子の立体配座について見てみようとMM2で計算してみたが当時のPC98では一晩に1配座という速度でしか計算ができなかった。それでも、適当に結合を回転させてその妥当性を確かめた。
最近のパソコンはとにかく速い。Spartanで1296配座をMMFF(分子力学)で配座解析したところ、30分もかからなかった。(そんな気がする。正確な時間を計っているわけではない)
そこで興味を持ったのが、「ほかの計算方法で同じ結果になるのであろうか」ということである。
Spartanにメモリーされたグローバルミニマムから100配座を初期配座として、SYBYL、AM1、PM3で再最適化計算してみた。ちなみにAM1、PM3は半経験的分子軌道法。時間がかかると覚悟したが、1配座当たり、最適化に3分強と100配座でも半日で終了した。パソコン恐るべきと感じた。
この計算ではMMFFで求めたローカルミニマムを初期配座としているので、同じ溝に落ちるはずであるが、やってみて愕然。計算によって求められるエネルギーがばらばらである。
下にはその結果をグラフにした。MMFFのエネルギーの順番でソートしたのでMMFFでは右上がりの線になっている。本来ならこの線に近いグラフを与えるはずなのに。。。???
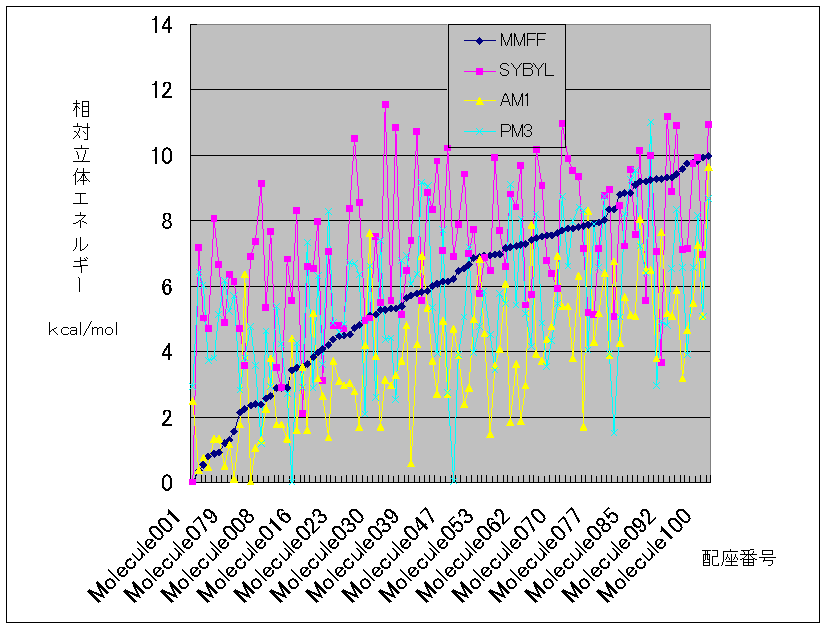
このグラフを見れば明らかなように、計算方法によって最安定構造はまちまちである。
下の構造はMMFF(左)およびAM1(右)で求められた最安定配座である。X線の構造はMMFFの算出した最安定配座に似ている。SYBYLで計算してもこの配座を初期配座にして再安定化させたものがグローバルミニマムとなっていた。しかし、半径検分子軌道法では、まったく相関が見られない。AM1での最適配座は二つの側鎖が離れた配座になっている。AM1ではMMFFのグローバルミニマムを初期配座にした構造は2.5kcal/molもエネルギーが高い構造になっている。これでは何が正しいのかさっぱりわからなくなってしまった。

計算は正しいと思った結果が出たところでやめるのが適当なのか?可能かはわからないが、ab initio法でもやってみようと思っている。