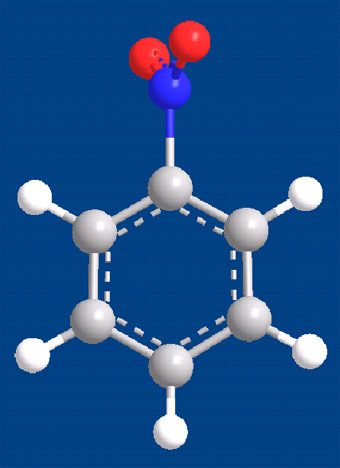
MINDO法について
平野氏の「分子軌道法MOPACガイドブック」(海文堂)によれば、MOPAC(version6)でMNDOをハミルトニアンとして使うと、ニトロベンゼンのニトロ基がベンゼン環に直交してしまうと書かれてあった。
ハミルトニアンが同じなら結果はほぼ同じであろうとSpartan04でMNDOを選択し、ニトロベンゼンを最適化してみた。予想通り結果は同じであった。Chem3DのCs MOPAC Pro (v8.0)で計算してみても結果は同じであった。やっぱりハミルトニアンは系全体のエネルギーを表す関数だと実感した。なお、ハミルトニアンの評価についてはFujitsuのホームページにもでているので参考になる。
http://venus.netlaboratory.com/material/messe/mopac2002/mopac_club/hamiltonian.html
もちろんPM3、AM1は平面分子を与えた。ついでと思い分子力学法でもやってみた。Spartan04のMMFF、SYBYLはどちらも平面分子を与えたが、Chem3DのMM2は下に示したような中途半端な分子になった。ニトロ基の配座については計算のたびに結果が違うので困ったもんだ。
Chem3Dが悪いのか、MNDOが悪いのか、それとも自分の操作が間違っているのかは不明。